2025
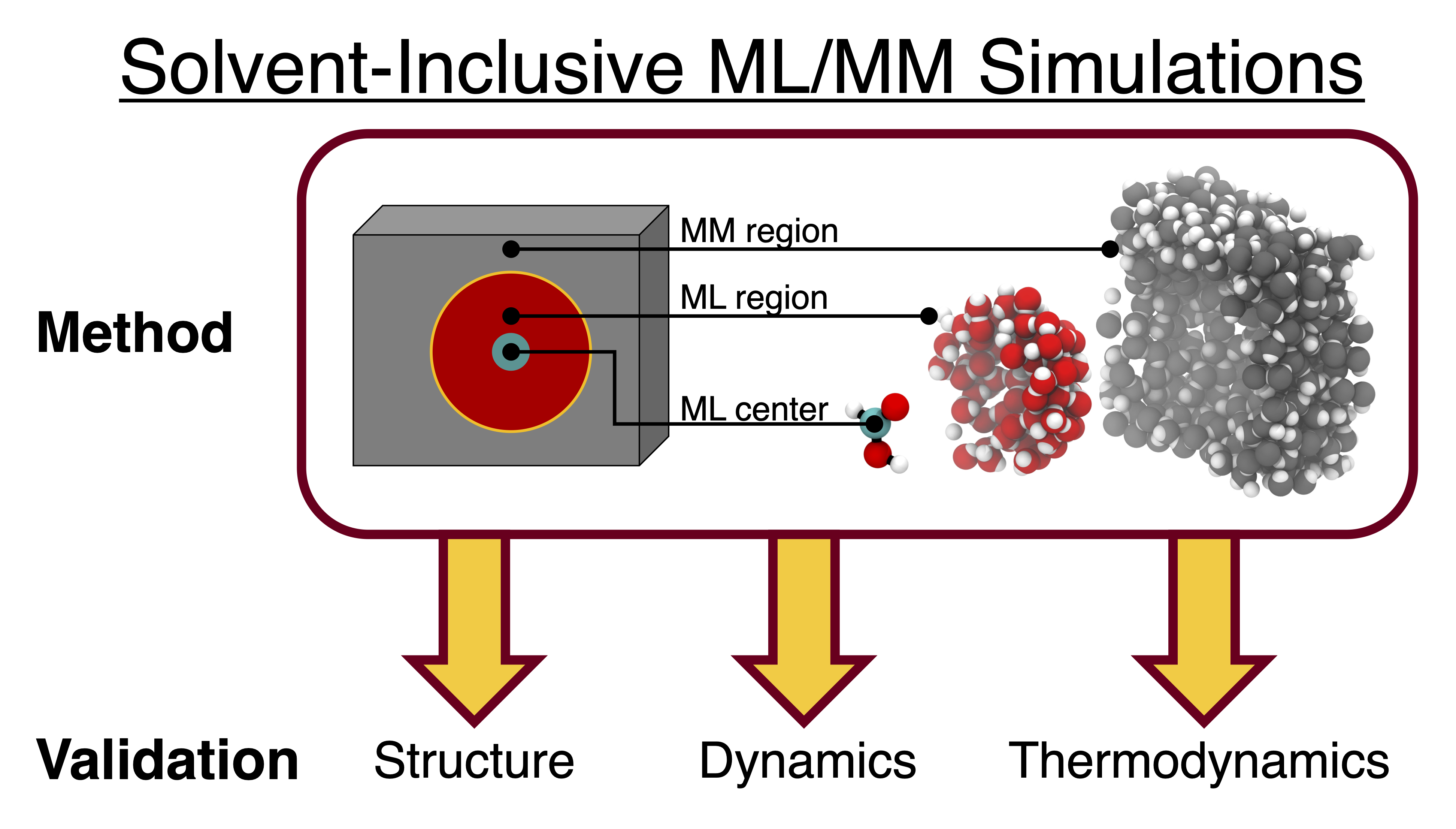
Solvent-Inclusive ML/MM Simulations: Assessments of Structural, Dynamical, and Thermodynamic Accuracy
Varun Gopal, Clara Kirkvold, Adrian Gordon, Jason Goodpaster, Sapna Sarupria. (2025) ChemRxiv.DOI: 10.26434/chemrxiv-2025-dlr6c
Crystal Nucleation Kinetics and Mechanism: Influence of Interaction Potential
Porhouy Minh, Steven W. Hall, Ryan S. DeFever, Sapna Sarupria. (2025) arXiv:2506.16541
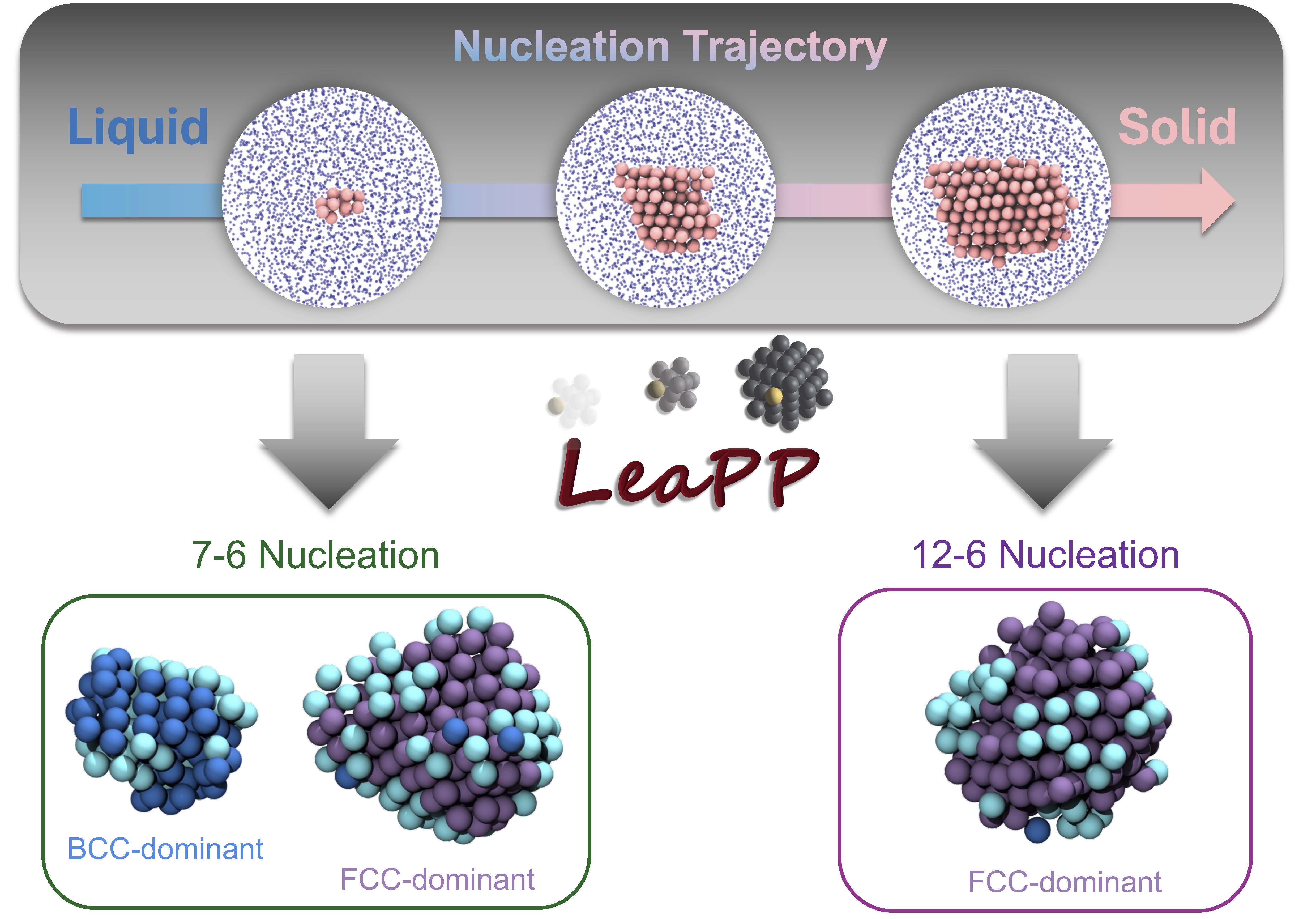
LeaPP: Learning Pathways to Polymorphs through Machine Learning Analysis of Atomic Trajectories
Steven W. Hall*, Porhouy Minh*, Sapna Sarupria. 21, 4121-4133, (2025) Journal of Chemical Theory and Computation.DOI: 10.1021/acs.jctc.5c00097
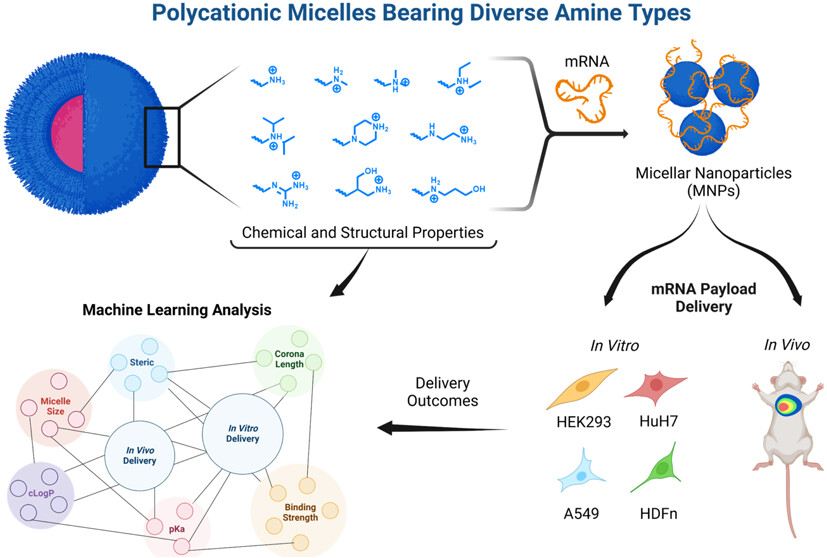
Machine Learning Reveals Amine Type in Polymer Micelles Determines mRNA Binding, In Vitro, and In Vivo Performance for Lung-Selective Delivery
Sidharth Panda, Ella J. Eaton, Praveen Muralikrishnan, Erin M. Stelljes, Davis Seelig, Michael C. Leyden, Alexandria K. Gilkey, Jackson T. Barnes, David V. Morrissey, Sapna Sarupria, Branden S. Moriarity, Theresa M. Reineke. 5, 1845-1861, (2025) JACS Au.DOI: 10.1021/jacsau.5c00084
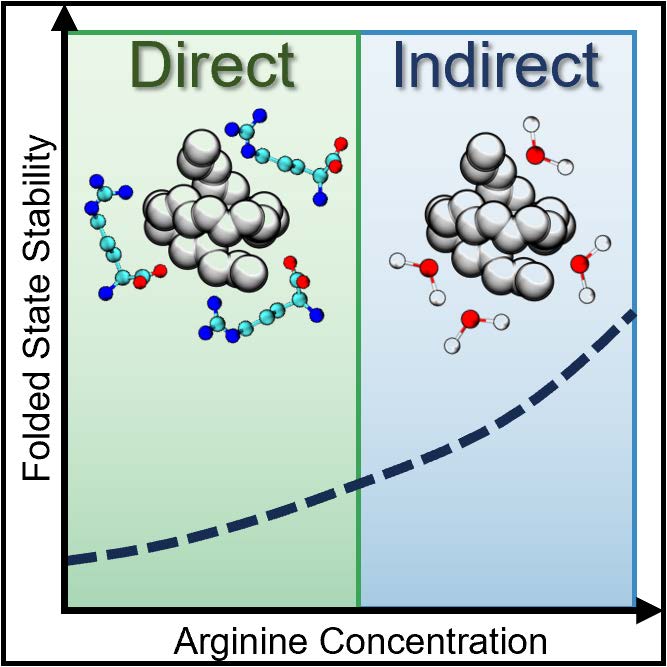
Flipping out: role of arginine in hydrophobic interactions and biological formulation design
Jonathan W. P. Zajac, Praveen Muralikrishnan, Idris Tohidian, Xianci Zeng, Caryn L. Heldt, Sarah L. Perry, Sapna Sarupria. 16, 6780-6792, (2025) Chemical Science.DOI: 10.1039/d4sc08672d

Towards Stable Biologics: Understanding Co-Excipient Effects on Hydrophobic Interactions and Solvent Network Integrity
Jonathan W. P. Zajac, Praveen Muralikrishnan, Caryn L. Heldt, Sarah L. Perry, Sapna Sarupria. 10, 432–446, (2025) Molecular Systems Design & Engineering.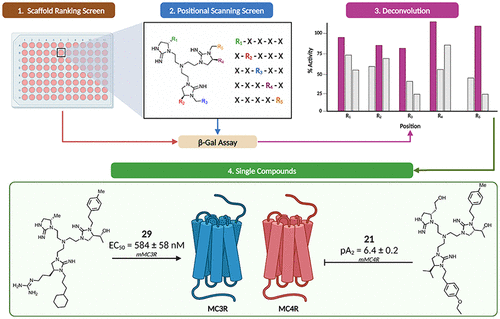
N-Branched Tricyclic Guanidines as Novel Melanocortin-3 Receptor Agonists and Melanocortin-4 Receptor Antagonists
Nicholas A. Weirath, Jonathan W. P. Zajac, Haley M. Donow, Travis M. Lavoi, Clemencia Pinilla, Radleigh G. Santos, Ritu Prajapati, Robert Speth, Mark D. Ericson, Sapna Sarupria, Marcello A. Giulianotti, Carrie Haskell-Luevano. 68, 2504−2527, (2025) Journal of Medicinal Chemistry.DOI: 10.1021/acs.jmedchem.4c01556
2023
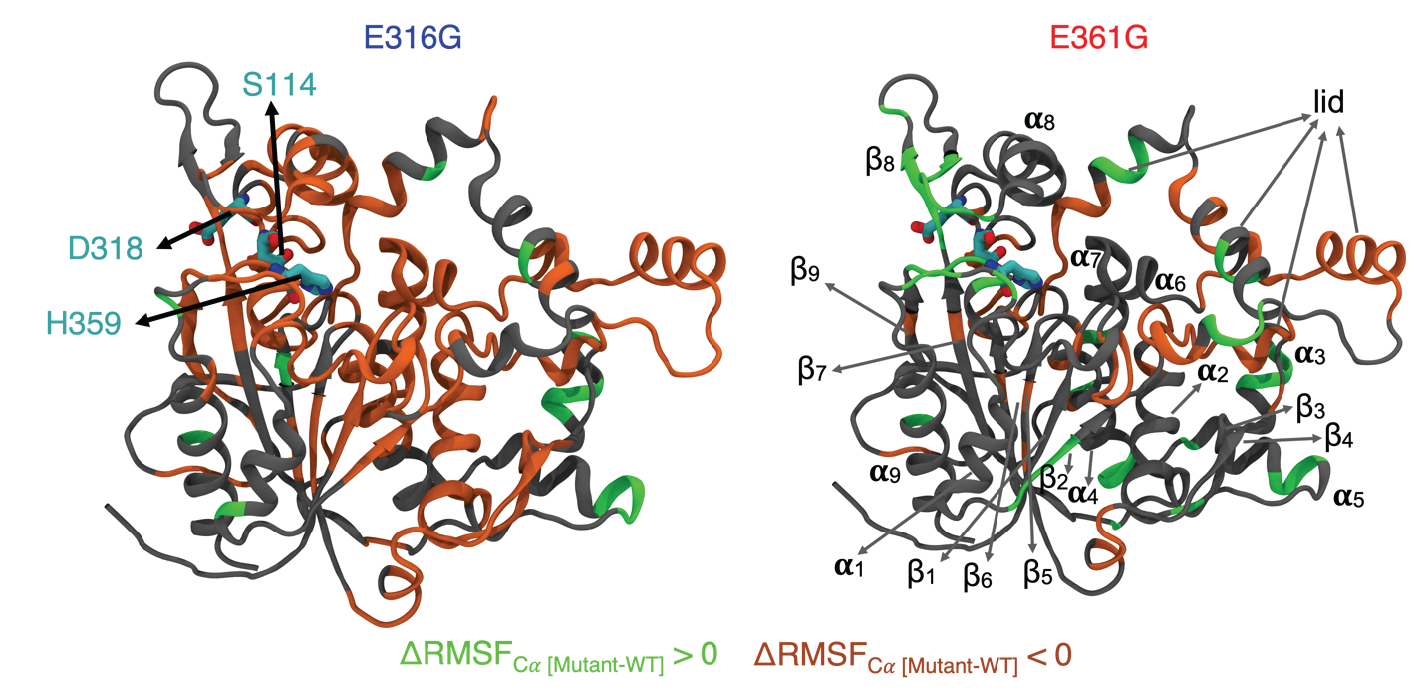
Exploitation of active site flexibility-low temperature activity relation for engineering broad range temperature active enzymes
Siva Dasetty, Jonathan W. P. Zajac and Sapna Sarupria. 8, 1355-1370, (2023) Molecular Systems Design & Engineering.DOI: 10.1039/D3ME00013C
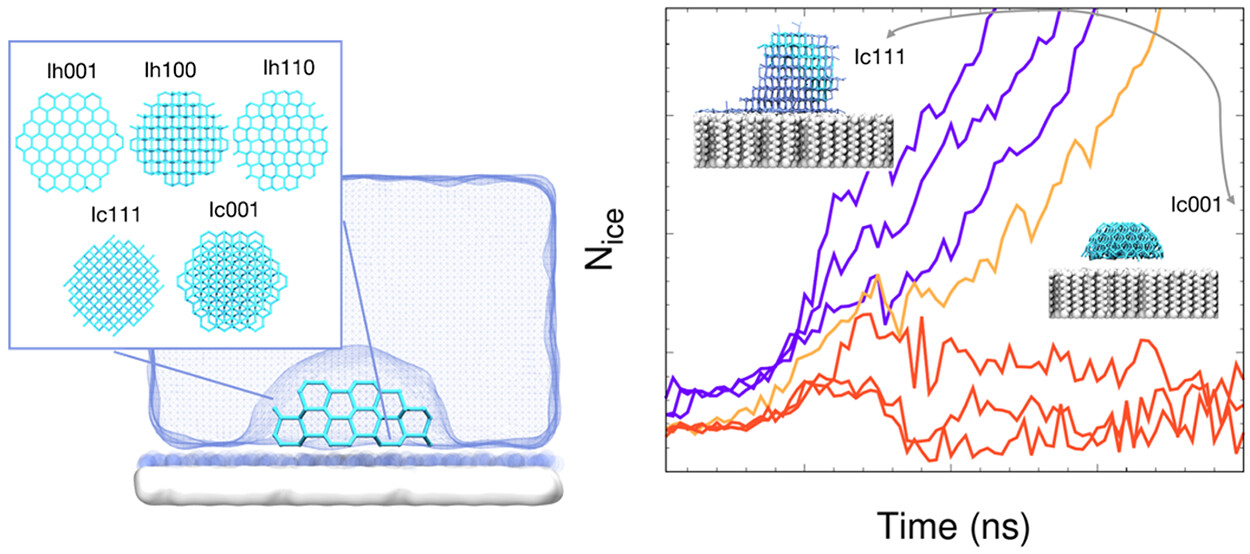
RSeeds: Rigid Seeding Method for Studying Heterogeneous Crystal Nucleation
Tianmu Yuan, Ryan S. DeFever, Jiarun Zhou, Ernesto Carlos Cortes-Morales, and Sapna Sarupria. 127, 4112–4125, (2023) The Journal of Physical Chemistry B.DOI: 10.1021/acs.jpcb.3c00910
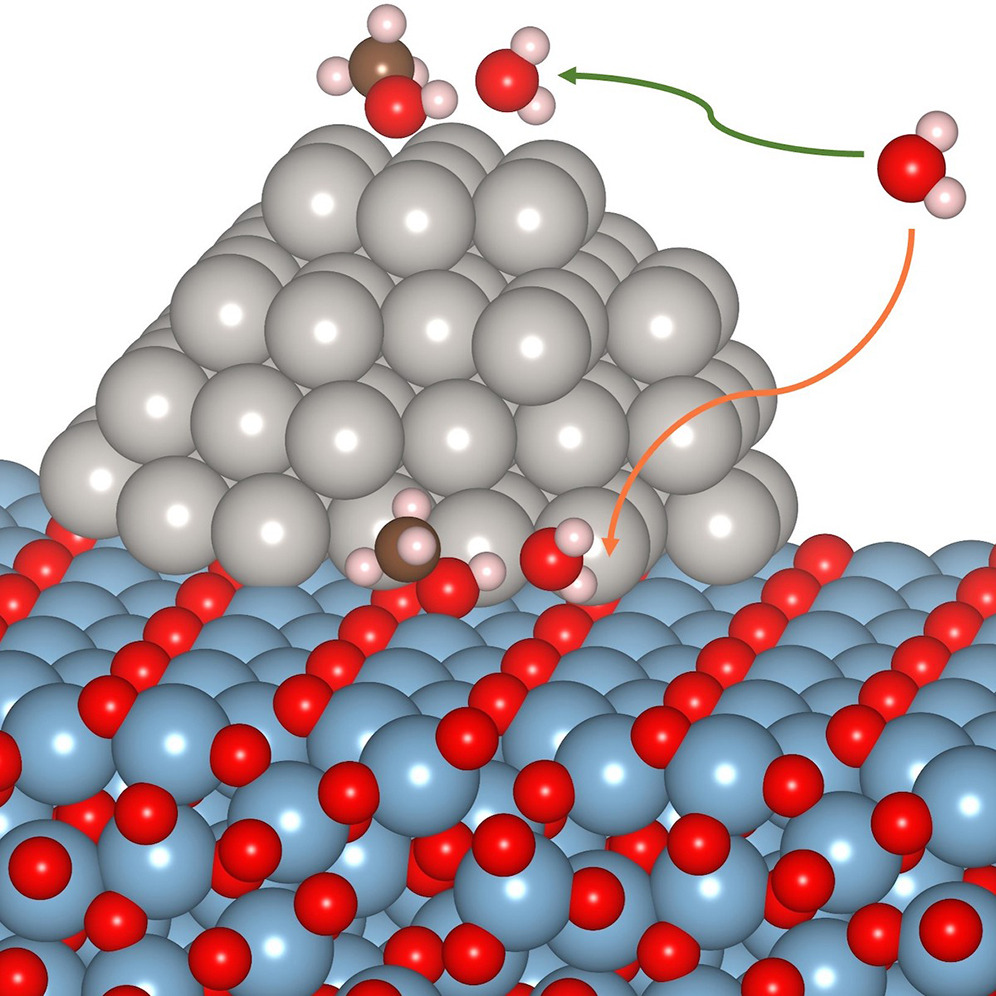
Differences in solvation thermodynamics of oxygenates at Pt/Al2O3 perimeter versus Pt (111) terrace sites
Ricardo A Garcia Carcamo, Xiaohong Zhang, Ali Estejab, Jiarun Zhou, Bryan J Hare, Carsten Sievers, Sapna Sarupria, Rachel B Getman. 26, 105980, (2023) iScience.DOI: 10.1016/j.isci.2023.105980
2022
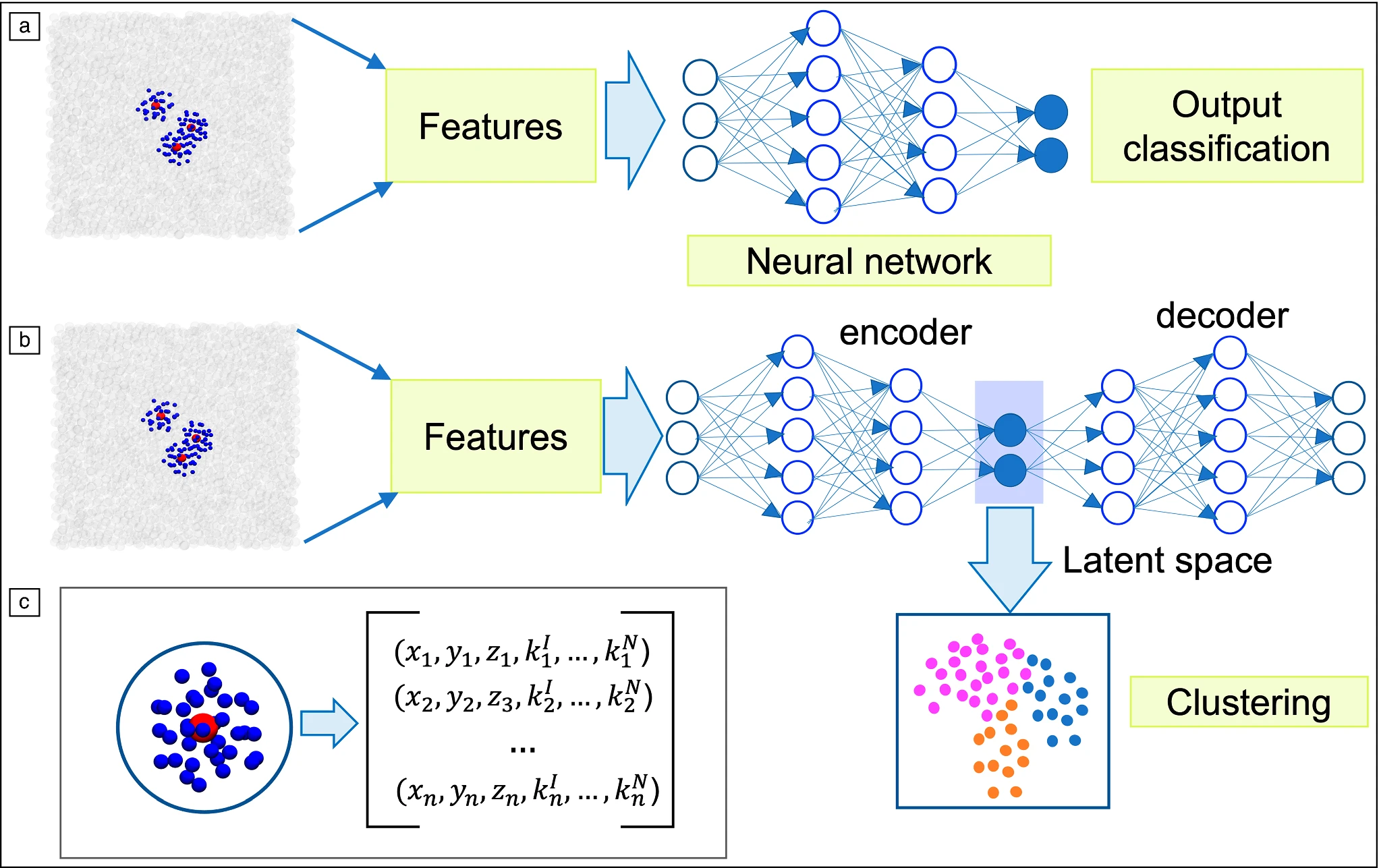
Machine learning for molecular simulations of crystal nucleation and growth
Sapna Sarupria, Steven W. Hall, Jutta Rogal. 47, (2022) MRS Bulletin.DOI: 10.1557/s43577-022-00407-1
Practical guide to replica exchange transition interface sampling and forward flux sampling
Steven W. Hall, Grisell Díaz Leines, Sapna Sarupria, Jutta Rogal. 156, 200901, (2022) The Journal of Chemical Physics.DOI: 10.1063/5.0080053
2021
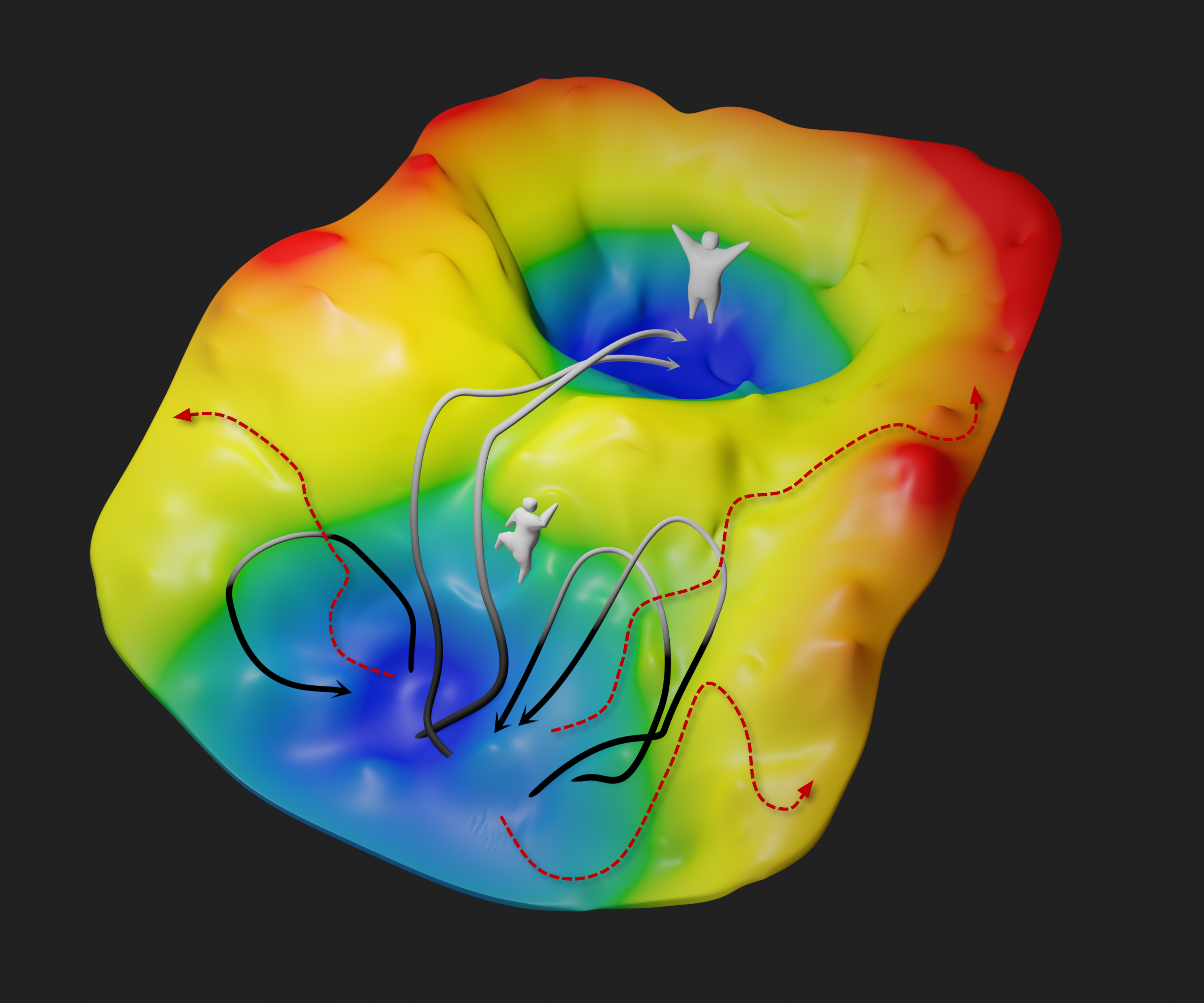
The Middle Science: Traversing Scale In Complex Many-Body Systems
Aurora E. Clark, Henry Adams, Rigoberto Hernandez, Anna I. Krylov, Anders M. N. Niklasson, Sapna Sarupria, Yusu Wang, Stefan M. Wild, and Qian Yang 7 1271-1287 (2021) ACS Central Science.DOI: 10.1021/acscentsci.1c00685
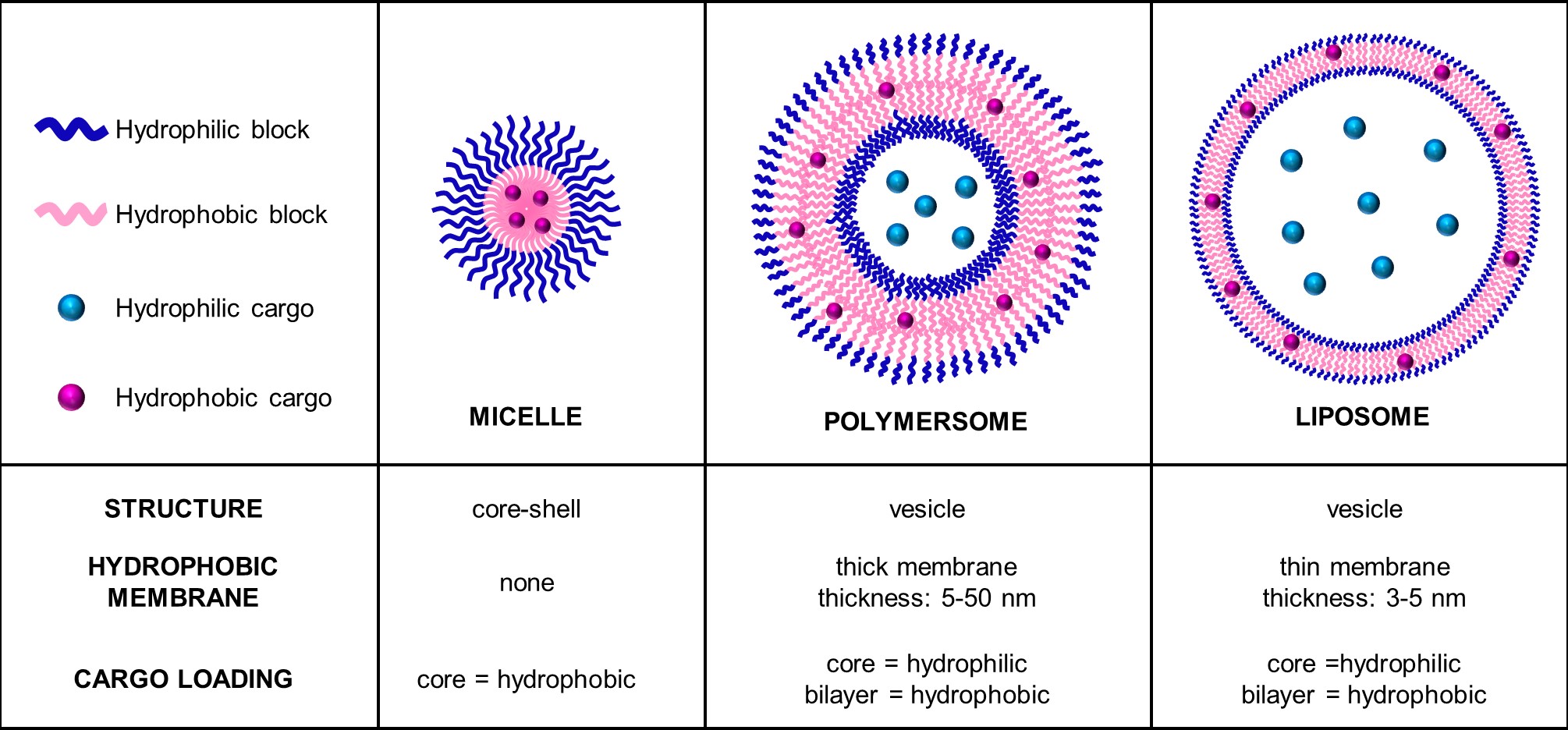
Toward enzyme-responsive polymersome drug delivery
Bipin Chakravarthy Paruchuri, Varun Gopal, Sapna Sarupria, and Jessica Larsen 16 2679-2693 (2021) Nanomedicine.DOI: 10.2217/nnm-2021-0194
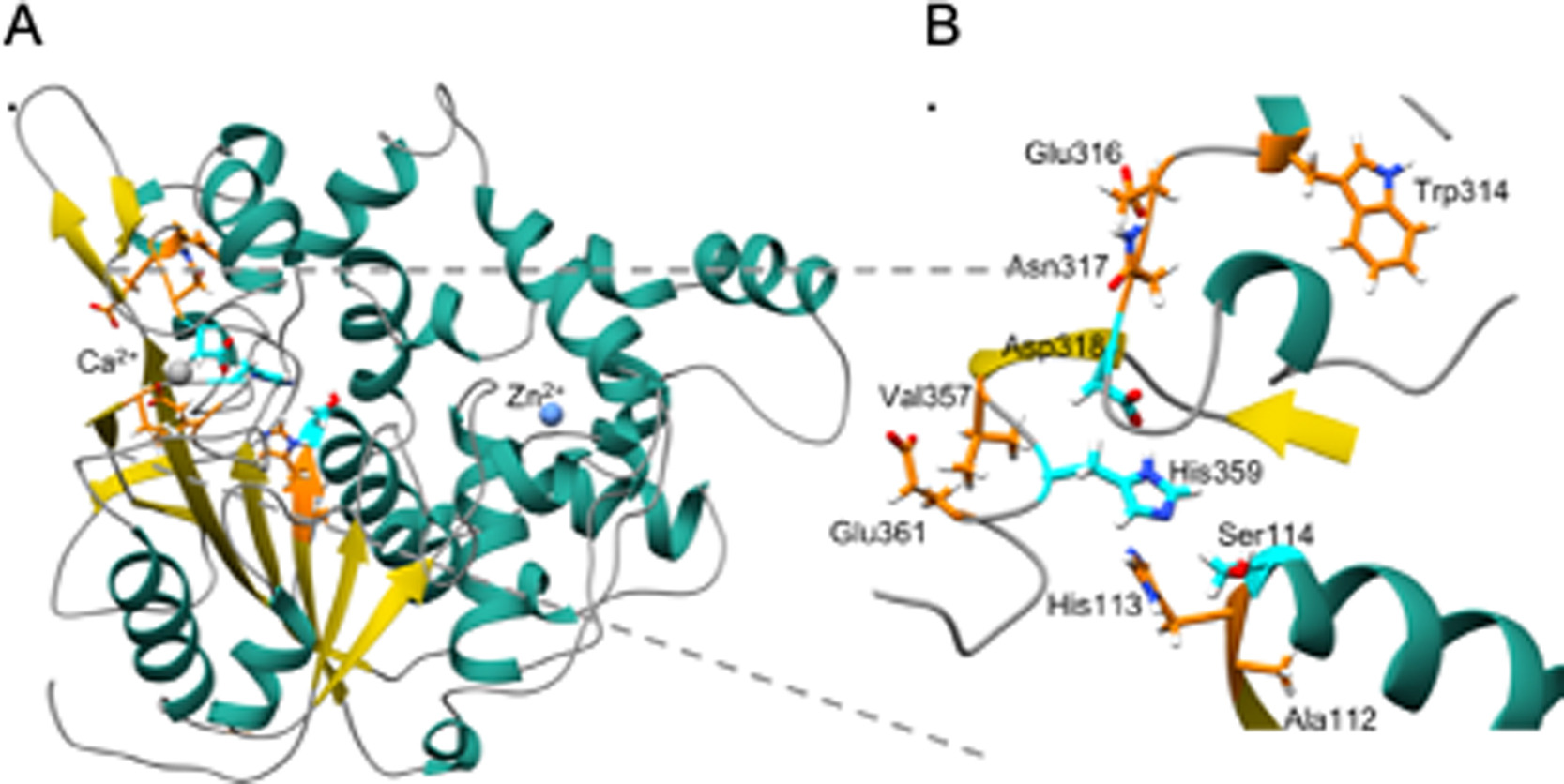
Rational engineering of low temperature activity in thermoalkalophilic Geobacillus thermocatenulatus lipase
Weigao Wang, Siva Dasetty, Sapna Sarupria, and Mark Blenner 174 Page N.A. (2021) Biochemical Engineering Journal.DOI: 10.1016/j.bej.2021.108093
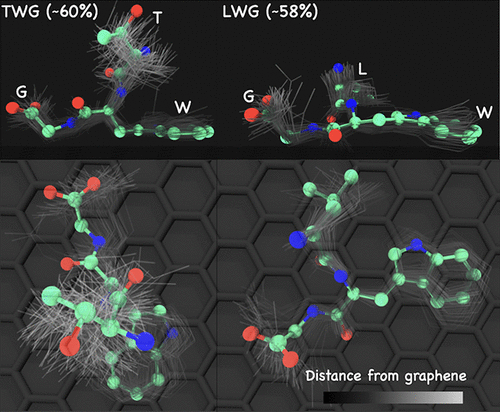
Advancing Rational Control of Peptide–Surface Complexes
Siva Dasetty and Sapna Sarupria 10 2644-2657 (2021) The Journal of Physical Chemistry B.DOI: 10.1021/acs.jpcb.0c10740
2020
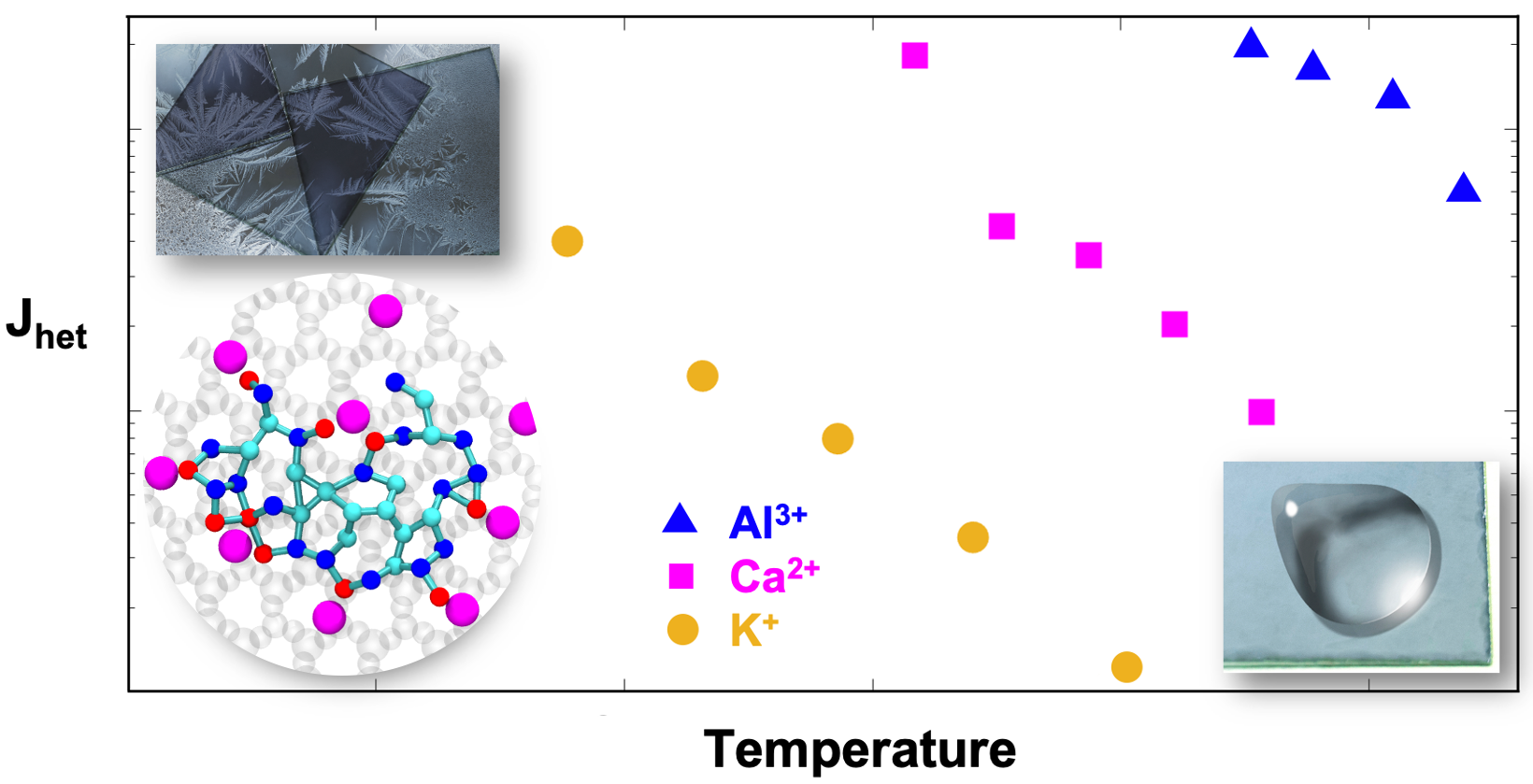
Multivalent Surface Cations Enhance Heterogeneous Freezing of Water on Muscovite Mica
Nurun Nahar Lata, Jiarun Zhou, Pearce Hamilton, Michael Larsen, Sapna Sarupria, and Will Cantrell 11 8682-8689 (2020) The Journal of Physical Chemistry Letters.DOI: 10.1021/acs.jpclett.0c02121
2019
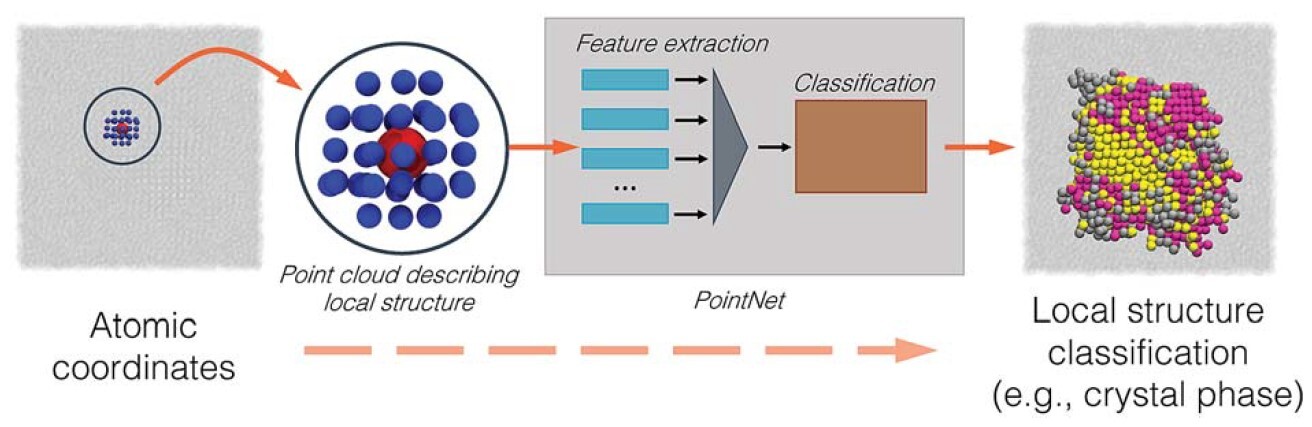
A generalized deep learning approach for local structure identification in molecular simulations
RS DeFever, C Targonski, SW Hall, MC Smith, and S Sarupria 10 7503-7515 (2019) Chemical Science.DOI: 10.1039/C9SC02097G
Building A Scalable Forward Flux Sampling Framework using Big Data and HPC
RS DeFever, W Hanger, J Kilgannon, A Apon, S Sarupria, and L Ngo (2019) Practice and Experience in Advanced Research Computing (PEARC19)DOI: 10.1145/3332186.3332205
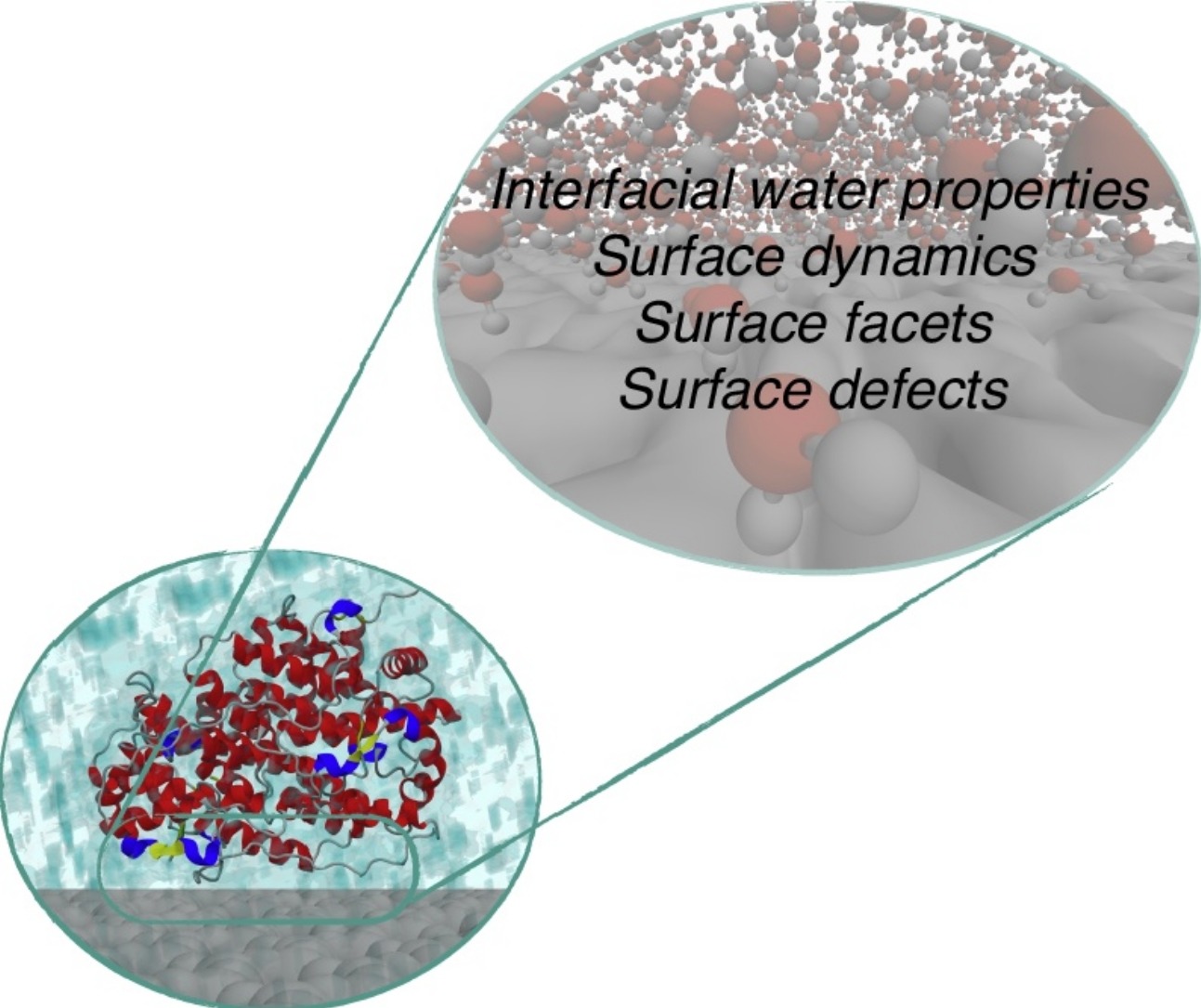
Simulations of interfacial processes: Recent advances in force field development
S Dasetty, P Meza-Morales, S Sarupria, and RB Getman 23 138-145 (2019) Current Opinion in Chemical Engineering.DOI: 10.1016/j.coche.2019.04.003
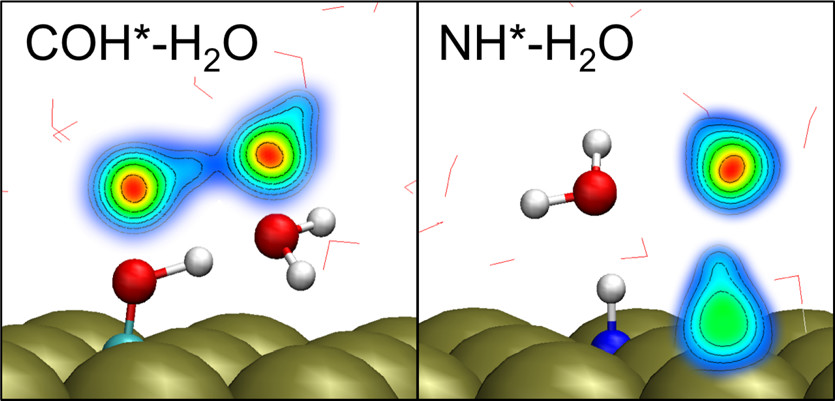
Free energies of catalytic species adsorbed to Pt(111) surfaces under liquid solvent calculated using classical and quantum approaches
X Zhang, RS DeFever, S Sarupria, and RB Getman 595 2190-2198 (2019) Journal of Chemical Information and Modeling.DOI: 10.1021/acs.jcim.9b00089
Multiscale Sampling of a Heterogeneous Water/Metal Catalyst Interface using Density Functional Theory and Force-Field Molecular Dynamics
CJ Bordenschatz, X Zhang, T. Xie, J Arvay, S Sarupria, and RB Getman 146 e59284 (2019) Journal of Visualized Experiments.DOI: 10.3791/59284-v
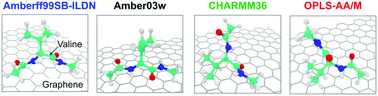
Adsorption of Amino Acids on Graphene: Assessment of Current Force Fields
S Dasetty, J Barrows, and S Sarupria 15 2359-2372 (2019) Soft Matter.DOI: 10.1039/C8SM02621A
Contour forward flux sampling: Sampling rare events along multiple collective variables
RS DeFever, S Sarupria 150 024103 (2019) Journal of Chemical Physics.DOI: 10.1063/1.5063358
2018
Introduction to the special issue on advanced molecular simulations: Methods and applications”, Editorial to Special Issue “Advanced molecular simulations: Methods and application
S Sarupria 17 (2018) Journal of Theoretical and Computational Chemistry.DOI: 10.1142/S0219633618020017

Surface chemistry effects on heterogeneous clathrate hydrate nucleation: A molecular dynamics study
RS DeFever, S Sarupria 117 205-213 (2018) Journal of Chemical Thermodynamics.DOI: 10.1016/j.jct.2017.08.021
2017
Nucleation mechanism of clathrate hydrates of water-soluble guest molecules
RS DeFever and S Sarupria 147 204503 (2017) Journal of Chemical Physics.DOI: 10.1063/1.4996132
Engineering Lipases: Walking the Fine Line Between Activity and Stability
S Dasetty, MA Blenner, and S Sarupria 4 113008 (2017) Materials Research Express.DOI: 10.1088/2053-1591/aa9946
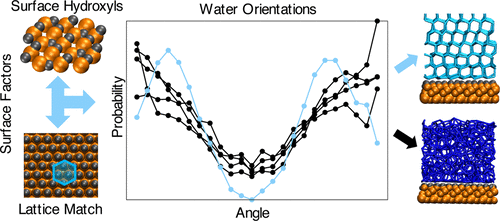
Heterogeneous ice nucleation: Interplay of surface properties and their impact on water orientations
B Glatz and S Sarupria 34 1190-1198 (2017) Langmuir.DOI: 10.1021/acs.langmuir.7b02859
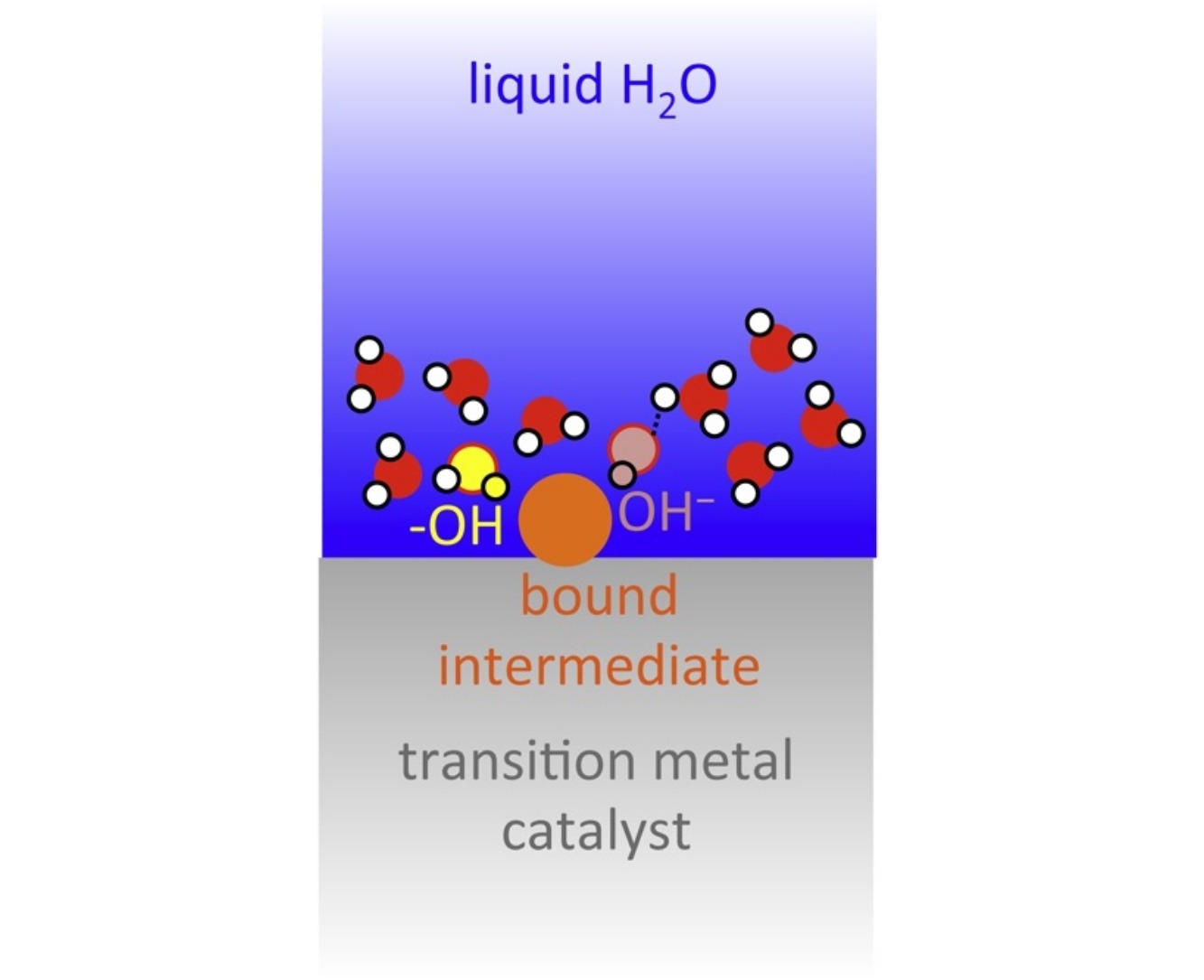
On the water structure at hydrophobic interfaces and the roles of water on transition-metal catalyzed reactions: A short review
X Zhang, TE Sewell, B Glatz, S Sarupria, and RB Getman 285 57-64 (2017) Catalysis Today.DOI: 10.1016/j.cattod.2017.02.002
A DFT and MD study of aqueous-phase dehydrogenation of glycerol on Pt(1 1 1): comparing chemical accuracy versus computational expense in different methods for calculating aqueous-phase system energies
T Xie, S Sarupria, and RB Getman 43 370-378 (2017) Molecular Simulation.DOI: 10.1080/08927022.2017.1285403
2016
The surface charge distribution affects the ice nucleating efficiency of silver iodide
B Glatz and S Sarupria 145 211924 (2016) Journal of Chemical Physics.DOI: 10.1063/1.4966018
2015
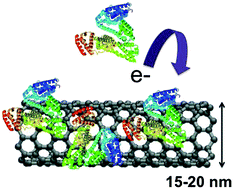
Influence of carbon nanomaterial defects on the formation of protein corona
B Sengupta, WE Gregory, J Zhu, S Dasetty, M Karakaya, JM Brown, AM Rao, JK Barrows, S Sarupria, and R Podilla 5 82395-82402 (2015) RSC Advances.DOI: 10.1039/C5RA15007H
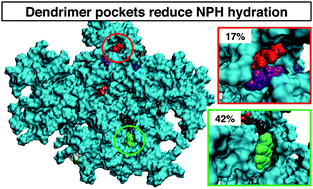
Association of small aromatic molecules with PAMAM dendrimers
RS DeFever and S Sarupria 17 29548-29557 (2015) Physical Chemistry Chemical Physics.DOI: 10.1039/C5CP03717D
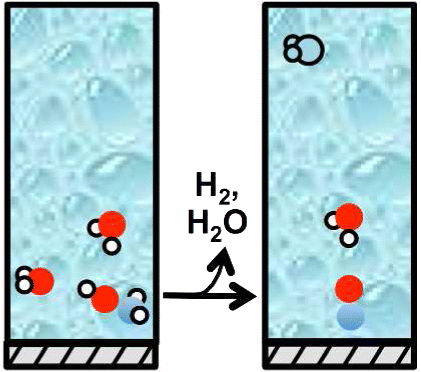
Molecular-Level Details about Liquid H2O Interactions with CO and Sugar Alcohol Adsorbates on Pt(111) Calculated Using Density Functional Theory and Molecular Dynamics
CJ Bordenschatz, S Sarupria, and RB Getman 119 13642-13651 (2015) Journal of Physical Chemistry C.DOI: 10.1021/acs.jpcc.5b02333
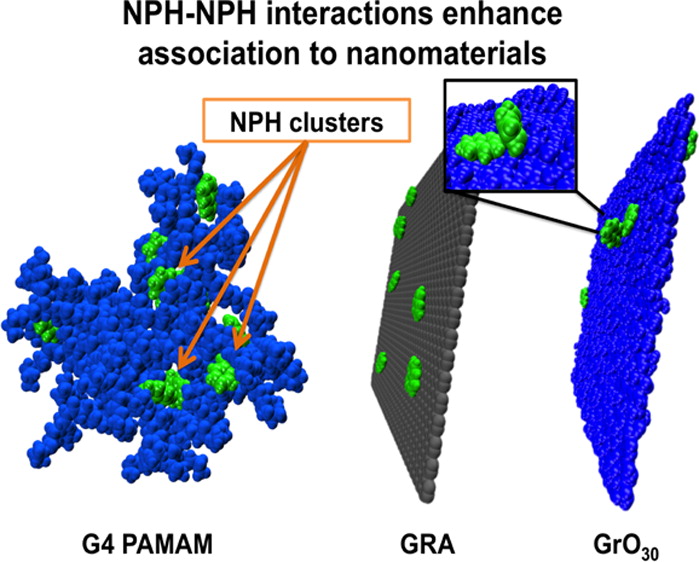
PAMAM dendrimers and graphene: Materials for removing aromatic contaminants from water
RS DeFever, NK Geitner, P Bhattacharya, F Ding, PC Ke, and S Sarupria 49 4490-4497 (2015) Environmental Science and Technology.DOI: 10.1021/es505518r
2014
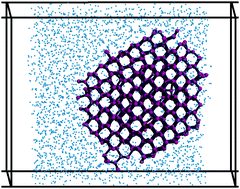
Suppression of sub-surface freezing in free-standing thin films of a coarse-grained model of water
A Haji-Akbari, RS DeFever, S Sarupria, and PG Debenedetti 16 25916-25927 (2014) Physical Chemistry Chemical Physics.DOI: 10.1039/C4CP03948C
2013
Molecular Dynamics Simulations of Peptide–SWCNT Interactions Related to Enzyme Conjugates for Biosensors and Biofuel Cells
O Karunwi, C Baldwin, G Griecheimer, S Sarupria, and A Guiseppi-Elie 3 (4) 1343007 (2013) Nano LIFE.DOI: 10.1142/S1793984413430071
SciFlow: A dataflow-driven model architecture for scientific computing using Hadoop
P Xuan, Y Zheng, S Sarupria, and A Apon 36-44 (2013) IEEE International Conference on Big Data.DOI: 10.1109/BigData.2013.6691725
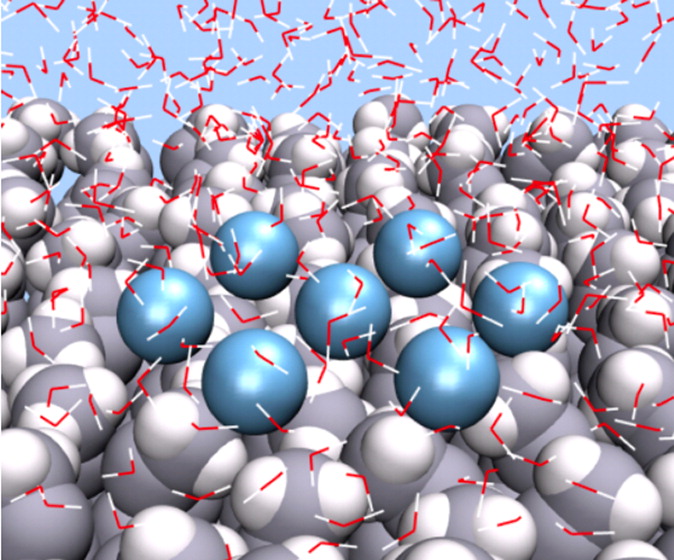
On the Thermodynamics and Kinetics of Hydrophobic Interactions at Interfaces
S Vembanur, AJ Patel, S Sarupria, and S Garde 117 (35) 10261-10270 (2013) Journal of Physical Chemistry B.DOI: 10.1021/jp4050513
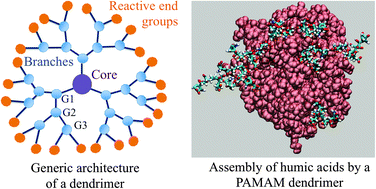
Exploiting the physicochemical properties of dendritic polymers for environmental and biological applications
P Bhattacharya, NK Geitner, S Sarupria, and PC Ke 15 (13) 4477-4490 (2013) Physical Chemistry Chemical Physics.DOI: 10.1039/C3CP44591G
2012
Homogeneous Nucleation of Methane Hydrate in Microsecond Molecular Dynamics Simulations
S Sarupria and PG Debenedetti 3 (20) 2942-2947 (2012) Journal of Physical Chemistry Letters.DOI: 10.1021/jz3012113
2011
Molecular Dynamics Study of Carbon Dioxide Hydrate Dissociation
S Sarupria and PG Debenedetti 115 (23) 6101-6111 (2011) Journal of Physical Chemistry A.DOI: 10.1021/jp110868t
2010
Studying pressure denaturation of a protein by molecular dynamics simulations
S Sarupria, T Ghosh, AE Garcia, and S Garde 78 (7) 1641-1651 (2010) Proteins.DOI: 10.1002/prot.22680
2009
Hydrate Molecular Ballet
PG Debenedetti and S Sarupria 326 (5956) 1070-1071 (2009) Science.DOI: 10.1126/science.1183027
Addition/Correction: Ion Pairing in Molecular Simulations of Aqueous Alkali Halide Solutions
CJ Fennell, A Bizjak, V Vlachy, KA Dill, S Sarupria, S Rajamani, and S Garde 113 (44) 14837-14838 (2009) Journal of Physical Chemistry B.DOI: 10.1021/jp908484v
Quantifying Water Density Fluctuations and Compressibility of Hydration Shells of Hydrophobic Solutes and Proteins
S Sarupria and S Garde 103 (3) 37803 (2009) Physical Review Letters.DOI: 10.1103/PhysRevLett.103.037803
2008
Enthalpy-Entropy Contributions to Salt and Osmolyte Effects on Molecular-Scale Hydrophobic Hydration and Interactions
MV Athawale, S Sarupria, and S Garde 112 (18) 5661-5670 (2008) Journal of Physical Chemistry B.DOI: 10.1021/jp073485n
2007
Pressure dependence of the compressibility of a micelle and a protein: insights from cavity formation analysis
B Pereira, S Jain, S Sarupria, L Yang, and S Garde 105 (2) 189-199 (2007) Molecular Physics.DOI: 10.1080/00268970601140750
