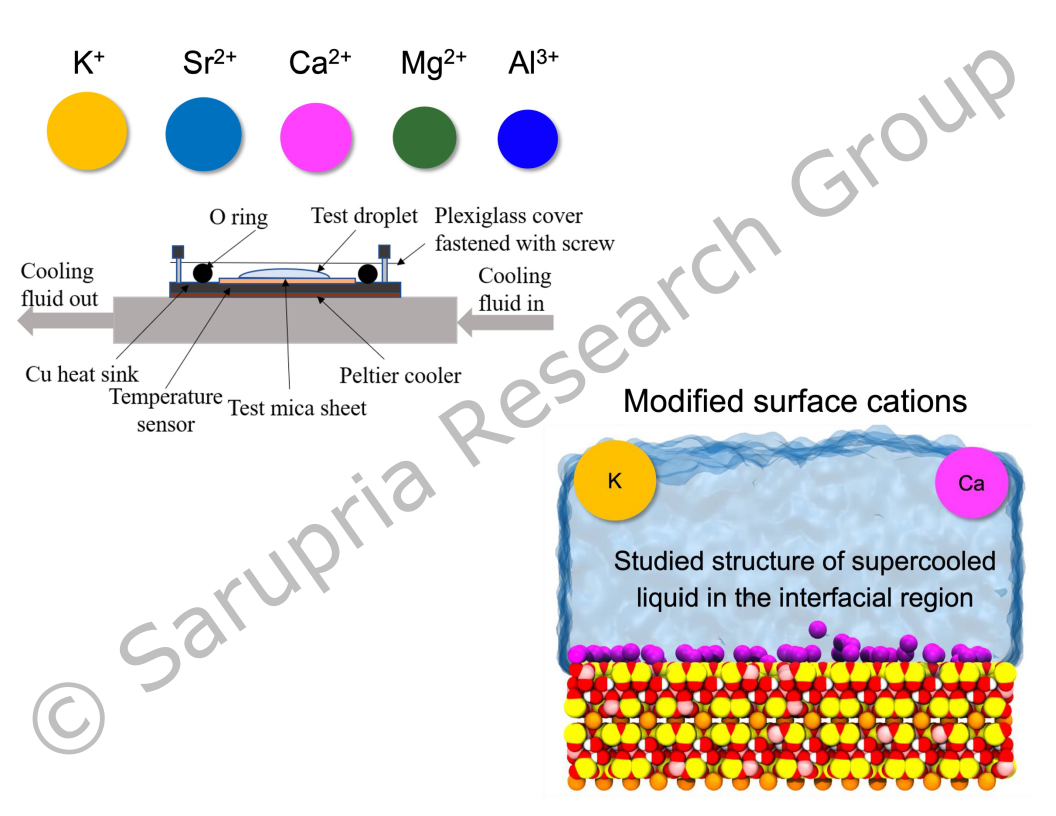
Heterogeneous ice nucleation is the nucleation process facilitated by the substrates, i.e. pollens, aerosol particles, and ice binding proteins. In the past few decades, researchers are working to answer the question of what makes a substrate a good/bad nucleating agent. In addition, how the corresponding nucleation mechanism changes from case to case also remains unsolved. Our group has investigated the ice nucleating behavior on several mineral surfaces with modified surface properties. We have identified the correlation between the metastable water structure and the surface nucleating propensity. We are working on studying the ice nucleation on mica surfaces with modified surface cations by combining the experimental techniques with computational methods. Our ultimate goal is to be able to engineer the surface with desired nucleating properties.
Check out our talk describing our latest work.
Our group develops methods and software in order to efficiently sample rare events in molecular simulations. Molecular processes such as nucleation from a metastable phase and protein folding involve waiting times many orders of magnitude longer than the actual transition to occur once it has begun. We develop and apply forward flux sampling-based methods that sample along multiple order parameters simultaneously as well as software—SAFFIRE—to enable their use in highly parallel computing environments.
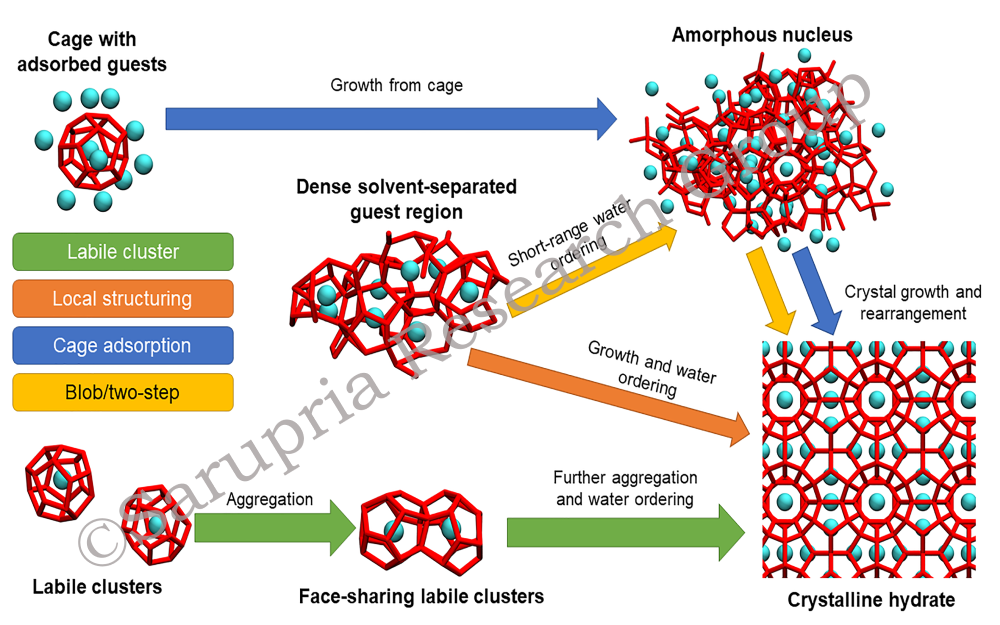
Aqueous mixtures of molecules like methane, carbon dioxide and ethane form beautiful crystalline structures called gas hydrates when pressurized and cooled. Naturally occurring methane gas hydrates are expected to be a promising source of energy. Further novel technologies to purify water, store energy, sequester carbon dioxide and separate gases have been based on gas hydrates. However, many molecular details on the formation of gas hydrates remain unknown. We use molecular simulations combined with rare event methods to answer these questions—what are the structural and dynamical pathways of hydrate nucleation, how is this transition affected by the presence of surfaces and how do additives affect such transitions?
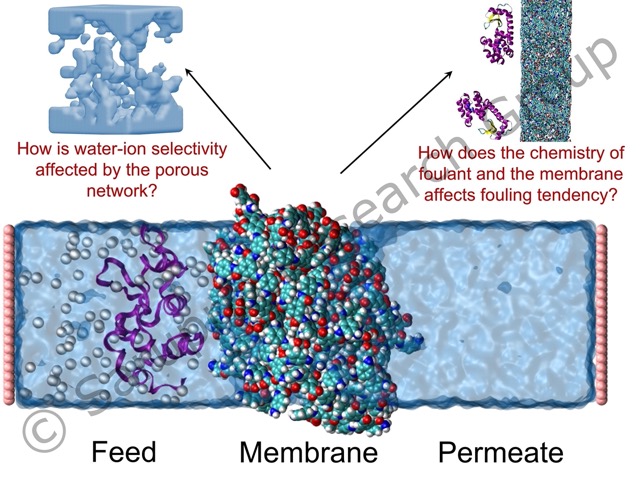
Desalination of seawater is touted to be an effective alternative to meet the drinking water demands in regions with stressed freshwater sources. Polyamide membranes are most widely used commercial desalination membranes for their superior salt rejection. However, the lack of understanding of precise rejection mechanism limits the ability to design highly selective membranes. The membranes with enhanced selectivity can help avoid costly remineralization for drinking water, and also selectively recover useful minerals from the seawater. Together with the group of Prof. Amir Haji-Akbari, we are using molecular simulations coupled with the forward flux sampling to determine the factors governing the water-ion selectivity.
Desalination is also an energy intensive process, and the membranes lose performance over time due to fouling. The propensity of a membrane to foul is strongly associated with the surface chemistry. Zwitterionic coatings on the membranes can reduce fouling due to the formation of a hydration layer. We are using molecular simulations coupled with the advanced sampling techniques, to determine the strength of the hydration layer for various zwitterionic moieties.
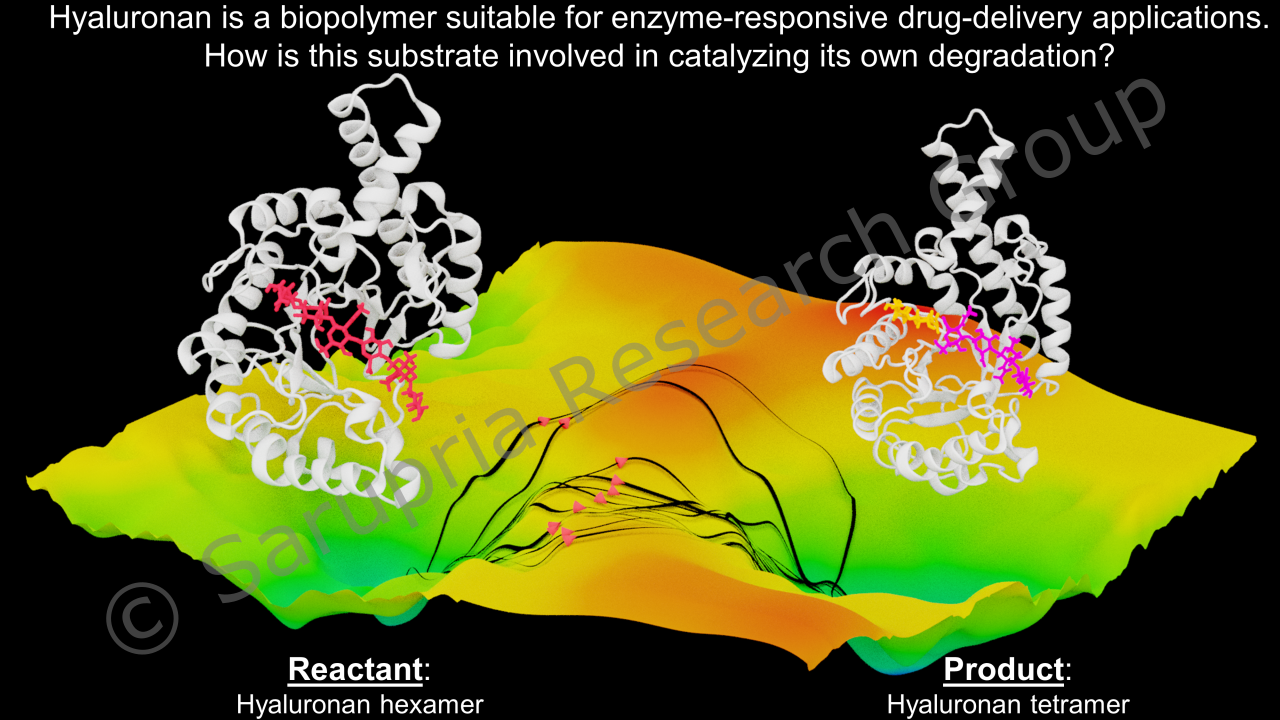
Enzymes are the proteins that catalyze reactions. The powerful quality of enzymes is their specificity. This has made them an attractive catalyst to use for biotechnological applications. However, enzymes can work only in certain conditions and often these conditions are different from those in the industrial applications. Thus, there is a need to engineer enzymes and enzyme environments such that the enzymes are functional in the desired operating conditions. To enable such technologies, we use molecular simulations to understand the structure-function-dynamics relation in enzymes. We study the effect of mutations as well as immobilization of enzymes on this relationship.
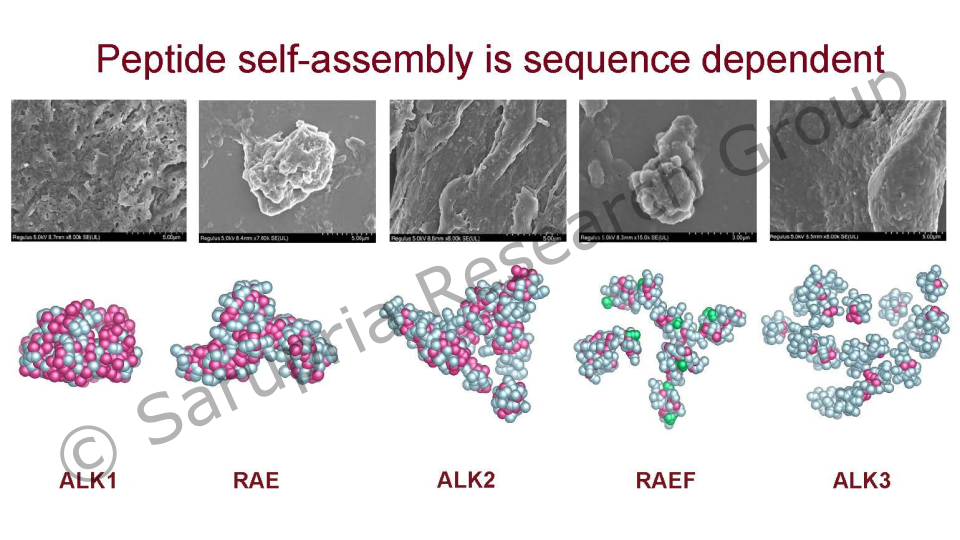
Peptide self-assembly is a process in which peptides spontaneously form ordered aggregates. Self-assembled short peptides have attracted widespread interest due to their tunable, biocompatible nature and have many promising applications, in energy materials, tissue engineering, sensing and drug delivery.
There are numerous interesting candidate molecules within the sequence space built from the 20 amino acids. Despite the ease of synthesis, the discovery of short peptides capable of self-assembly becomes a challenging problem to investigate experimentally due to the vast sequence space compounds (8E3 tripeptides to 6.4E7 hexapeptides). Thus, one needs methods to screen and predict the supramolecular behavior of peptides.
We are investigating the self-assembly of (short) peptides with a focus on hydrogel formation. The peptide self-assembly process is investigated at various levels of detail, from atomistic simulations to coarse-grained molecular dynamics simulations. Our approach also combines MD simulation and machine (active) learning methods to accelerate the design of peptides that form the desired supramolecular assemblies. The work leads to the identification and development of peptides that can be used for drug delivery.
This project maintains the experimental collaboration with Dr. Angela Alexander-Bryant and Dr. Leah B Casabianca at Clemson University. Dr. Casabianca's group utilizes solution and solid state NMR for biophysical characterization of peptide self-assembly. Dr. Alexander-Bryant's lab focuses on the engineering and development of biomaterials for gene therapies and drug delivery.

The primary method for viral vaccine stabilization is via storing formulations at cold temperatures. Many vaccines rely on the cold chain for production, storage, and transportation. Difficulties in maintaining the cold chain limits the availability of vaccines, particularly when transportation is required to deliver vaccines to other countries. Therefore, providing vaccines in a manner that is temperature-stable would eliminate the requirement of the cold chain, improving both the stability of the vaccine supply and the overall accessibility of vaccines. The most common method for stabilizing vaccine formulations is by the addition of excipients. However, there is a lack of understanding of the stabilizing mechanisms at play. As such, our group is combining molecular dynamics and machine learning to identify the molecular features of excipient mixtures that impart temperature stability in vaccine formulations.
